Next-generation sequencing

Overview
Next-generation sequencing (NGS) technology enables the investigation of genetic variants across the genome (whole-genome sequencing, GS) or targeted region(s) (e.g., whole exome sequencing, ES). DNA-based analysis in NGS platforms can be performed on different specimen types (e.g. chorionic villi, amniotic fluids, cord blood, saliva, peripheral blood and more). While other traditional genetic tests such as G-banded chromosome analysis (karyotyping) and chromosomal microarray analysis (CMA) detects only a subset of genomic changes in a patient’s DNA, GS is a single test for comprehensive detection of genetic variants and provides increased diagnostic yield of genetic disorders in prenatal/postnatal/reproductive genetic testing (PMID: 32451733, 31475041).
Currently, we provide four different DNA-based NGS genetic diagnosis_services, including:
FetalSeq
FetalSeq is a genetic test utilizing low-pass (low-coverage but high-throughput) whole-genome sequencing (LP-WGS) with in-house developed bioinformatic pipelines for identifying clinically significant copy-number variants (CNVs) and chromosome aneuploidies. FetalSeq is developed and validated by The Chinese University of Hong Kong. FetalSeq provides comprehensive genetic investigation across different applications, including miscarriages, invasive prenatal diagnosis for high risk pregnancies such as fetal ultrasound anomalies and/or other indications, and postnatal cases with developmental delay, intellectual disability, autism spectrum disorders or congenital anomalies (PMIDs: 26820068,28696555,31447483 & 32451733). FetalSeq detects virtually all known microdeletion and microduplication syndromes as well as copy number changes greater than 50kb with clinically significant exonic changes in human genome. It has a higher genome-wide resolution compared to CNV-seq (PMID: 24998187).
Method: Genomic DNA (50-100ng) extracted from the biopsied sample is subjected to non-target-enriched library construction. The sample is then sequenced on next-generation sequencing platforms including NovaSeq, HiSeq X Ten and BGISEQ-500/MGISEQ-2000. Unless otherwise indicated, approximately 0.25-fold read-depth (~15 million reads with single-end 50bp read length) is generated for each case. Sequencing reads are aligned to the human reference sequence (GRCh37/hg19) using the Burrows-Wheeler Alignment tool (BWA) (PMID: 19451168) and are classified into sliding windows (50kb with 5kb increments) in terms of the aligned coordinates. After GC-correction and population-based normalization, CNV detection is performed by our in-house bioinformatic pipeline (Increment Rate of Coverage). Data interpretation is carried out by referencing the medical literature and online databases according to the guidelines of the American College of Medical Genetics and Genomics.
Limitations: FetalSeq does not exclude all small chromosomal anomalies (<50kb), copy number polymorphisms, and chromosomal rearrangements in the human genome (PMID: 15286789). FetalSeq does not detect triploidy, uniparental disomy, low-level mosaicism (<30%), trinucleotide repeat expansions, balanced structural variations, indels or point mutations.
Turn Around Time (TAT): 10 working days
Specimen requirement:
| Sample type | Amount |
|---|---|
| Blood | 2-3 ml in EDTA bottles |
| Chorionic villi | 5 mg villi in transport medium |
| Amniotic fluid | >10 ml in sterile container |
| Placental tissues | At least 0.5ug minimal concentration of 30ng/uL; A260/A280 of ~1.8 |

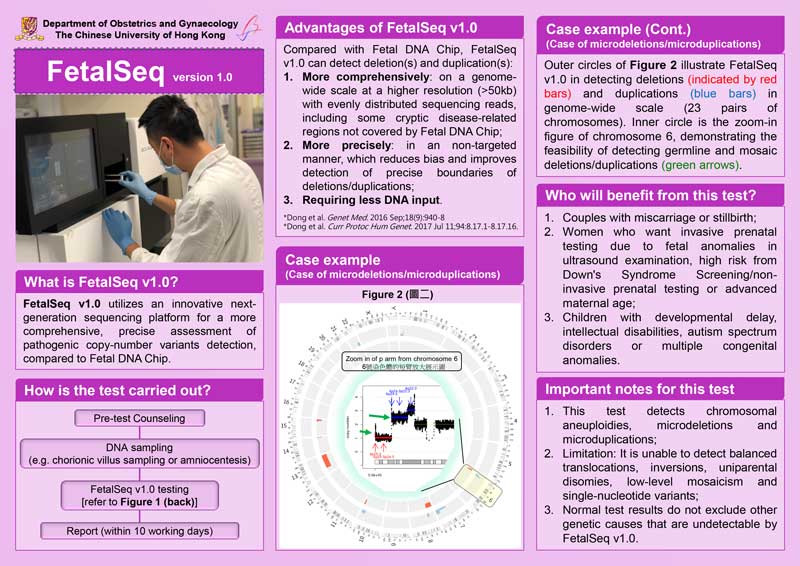
Figure (above) shows the robustness of FetalSeq v1.0 for detecting deletions (indicated by red bars) and duplications (blue bars) in genome-wide (24-chromosome) scale (outer circles). Zoom-in figure of chromosome 6 (shown in inner circle) demonstrates the feasibility of detecting constitutional and mosaic deletion/duplication (shown with green arrows).
ChromoSeq
ChromoSeq is a genetic test based on mate-pair (DNA fragment size ~5kb) low-pass whole-genome sequencing (LP-WGS) with in-house developed bioinformatic pipelines. ChromoSeq is developed and validated by The Chinese University of Hong Kong (PMIDs: 24610732,29095815,29364520, 31173071 & 31679651) in prenatal and postnatal genetic diagnosis as well as couples with recurrent miscarriages. In addition to chromosome aneuploidies (e.g. Down syndrome) and copy-number changes (e.g. DiGeorge Syndrome), ChromoSeq detects additional chromosomal structural rearrangements such as balanced translocations, inversions, insertions and complex rearrangements. In addition, it also identifies regions with absence of heterozygosity and uniparental isodisomy, which can be the cause of imprinting-related disorders (e.g. Beckwith Wiedemann syndrome, Russell-Silver syndrome).
ChromoSeq detects (1) virtually all known microdeletion and microduplication syndromes as well as copy number changes greater than 50kb with clinically significant exonic changes in human genome; (2) structural rearrangements such as balanced translocations, inversions, insertions and complex structural rearrangements; and (3) AOHs at a resolution of 5Mb. ChromoSeq is an enhanced edition of FetalSeq.
Method: Genomic DNA (2ug) extracted from the biopsied sample is subjected to an in-house mate-pair library construction method (CP-AL) using large DNA fragments (~5kb, PMID: 31173071). The library is sequenced on next-generation sequencing platforms including NovaSeq, HiSeq X Ten and MGISEQ-2000. Approximately 3-fold read-depth (~50 million read-pairs with paired-end 100bp in size) is generated for each case unless otherwise indicated. Sequencing reads are aligned to a reference sequence (GRCh37/hg19) using the Burrows-Wheeler Alignment tool (BWA) (PMID: 19451168). (1) For CNV analysis, reads are classified into sliding windows (50kb with 5kb increment) in terms of the aligned coordinates. After GC-correction and population-based normalization, CNV detection is performed by an in-house pipeline (Increment Rate of Coverage) and interpretation is carried out by referencing the medical literature and online databases according to the guidelines of the American College of Medical Genetics and Genomics. (2) For analysis of structural rearrangements, uniquely aligned read-pairs are further processed based on a four-step procedure: event clustering, systematic error filtering, random error filtering and alignment orientation. (3) For AOH detection, genotyping is carried out by analysis of mpileup file (Samtools). Regions (>5Mb) with a decreased rate of heterozygous SNVs and an increased rate of homozygous SNVs are detected as regions with AOH.
Limitations: ChromoSeq does not exclude all small chromosome anomalies (<50Kb), copy number polymorphisms. A normal ChromoSeq result does not exclude the possibility of triploidy, trinucleotide repeat expansions, low-level mosaicism (<30%), indels, or point mutation.
Turn Around Time (TAT): 3 weeks
Specimen requirement: Please contact our lab at (852) 3505 1557!
FAQ
Q1. Since ChromoSeq can detect balanced translocation, there is no more need to send parental blood for karyotyping, right?
Response: Yes, ChromoSeq is superior to karyotyping in identification of various complex chromosomal structural variants including balanced translocation, inversions as well microdeletions/duplications. However, the limitation of ChromoSeq remains in detecting Robertsonian translocation. Therefore, if there is recurrent trisomy including those acrocentric chromosomes (13, 14, 15, 21, 22) detected in the products of conception, parental karyotyping is still warranted to rule out Robertsonian translocation.
Q2. For recurrent miscarriage patient, should I send products of gestation (POG) for ChromoSeq first (no need to send parental sample to start with, right)?
Response: We do prefer to perform ChromoSeq for the POG together with the parental samples. Certainly, we can test the POG first for aneuploidies, genome-wide pathogenic copy-number variants (CNVs), structural rearrangements (SVs) and absence of heterozygosity (AOH, such as uniparental diasomy, UPD). The purpose of applying ChromoSeq in trio setting (with parental samples together) is for interpretation of any clinically significant variants detected by determination of the parental inheritance. Similar to the findings in CMA or FetalSeq, ChromoSeq identifies cryptic events (CNVs, SVs and AOHs) in the proband/fetus. It warrants the information of parental inheritance of each variant for further clinical interpretation.
Q3. What is the point of doing ChromoSeq + karyotyping?
Response: As explained in Q1, the limitation of ChromoSeq is in detection of Robertsonian translocation. To comprehensively identifying the genetic background of parental samples, it warrants a bundle of two tests. This is why we offer a lower price as package.
Q4. If I send you products of gestation and your team fail to find_ villi, what would be the charge?
Response: We will only charge the handling fee at HKD500. However, if karyotyping is also ordered, there will be another HKD1,000 charged.


GenomSeq
GenomSeq is one of the most powerful genomic tests which sequences nearly the entire genome at a high read-depth. Empowered by NGS technology and our in-house bioinformatics pipelines, GenomSeq comprehensively identifies genomic variants including single-nucleotide variants (SNVs), small insertions and deletions (InDels), copy-number variants (CNVs), structural rearrangements (SVs), and absence of heterozygosity (AOHs). It is developed and validated by The Chinese University of Hong Kong. GenomSeq’s applications include prenatal and postnatal genetic diagnosis (PMID: 31475041). The test detects (1) clinically significant SNVs and InDels; (2) virtually all known microdeletion and microduplication syndromes as well as copy number changes greater than 50kb with clinically significant exonic changes in human genome; (3) structural rearrangements such as balanced translocation, inversions, insertions, and complex rearrangements; and (4) AOHs at a resolution of 5Mb (recommended by the American College of Medical Genetics and Genomics, ACMG; PMID: 32296163). Patients have the option to opt-out of reporting of secondary findings and carrier status for disease-causing genes.
Method: Genomic DNA (500-1000ng) extracted from the biopsied specimen is subjected to a non-target-enriched PCR-free library construction. The library is sequenced on NGS platforms including NovaSeq, HiSeq X Ten and BGISEQ-500/MGISEQ-2000. A minimal of 30X or 30-fold read-depth (>300 million read-pairs with 150-bp read length) are generated for each case. Reads are aligned to the reference sequence (GRCh37/hg19) using the Burrows-Wheeler Alignment tool (BWA) (PMID: 19451168). (1) SNVs calling is carried out by HaplotypeCaller (GATK) and annotated by ANNOVAR (PMID: 20601685) with different public referenced databases. SNV interpretation is performed by referencing the medical literature and online databases according to the guidelines of the ACMG. (2) For CNV analysis, sequence reads are classified into sliding windows (50kb with 5kb increments). After GC-correction and population-based normalization, CNV detection is performed by an in-house pipeline (Increment Rate of Coverage) and interpretation is carried out by referencing the medical literature and online databases according to the guidelines of the American College of Medical Genetics and Genomics. (3) For analysis of structural rearrangements, uniquely aligned read-pairs are further processed based on a four-step procedure: event clustering, systematic error filtering, random error filtering and alignment orientation. (4) For AOH detection, genotyping is carried out by analysis of mpileup file (Samtools), and regions (>5Mb) with decreased rate of heterozygous SNVs and increased rate of homozygous SNVs are candidate AOHs.
Limitations: GenomSeq does not exclude all small chromosomal anomalies such as trinucleotide repeat expansions, triploidy, and low-level mosaicism (<30%). Mutations in some genes cannot be detected due to technical reasons including local sequence characteristics or the presence of closely related pseudogenes.
Turn Around Time (TAT): 20 calendar days (Trio), 28 calendar days (Proband only).
Specimen requirement: Please contact our lab at (852) 3505 1557.
FetalExome
FetalExome is a genetic test that analyzes and identifies known and potential disease-causing single nucleotide variants (SNVs) and small insertions and deletions (InDels) in nearly the entire protein-coding region of the genome (namely exome). In addition, FetalExome can also detect genome-wide absence of heterozygosity (AOH). FetalExome is ordered when prenatal ultrasound imaging detects an anomaly that strongly suggests that the fetal abnormality is caused by a monogenic disorder (by SNV or InDel). FetalExome is recommended to be performed in a trio setting and is often considered after a negative finding from chromosomal microarray analysis (CMA). Patients have the option to opt-out of reporting of secondary findings and carrier status for disease-causing genes.
Method: Genomic DNA (500-1000ng) extracted from the biopsied specimen is subjected to a non-target-enriched PCR-free library construction. The library is sequenced on NGS platforms including NovaSeq, HiSeq X Ten and BGISEQ-500/MGISEQ-2000. A minimal of 30X or 30-fold read-depth (~300 million read-pairs with 150-bp read length) are generated for each case. Reads are aligned to the reference sequence (GRCh37/hg19) using the Burrows-Wheeler Alignment tool (BWA) (PMID: 19451168). (1) SNVs calling is carried out by HaplotypeCaller (GATK) and annotated by ANNOVAR (PMID: 20601685) with different referenced databases. The analysis, interpretation, and reporting of variants will be confined to the exome. SNV interpretation is performed by referencing the medical literature and online databases according to the guidelines of the American College of Medical Genetics and Genomics. (2) For AOH detection, genotyping is carried out by analysis of mpileup file (Samtools), and regions (>5Mb as recommended by the American College of Medical Genetics and Genomics, ACMG; PMID: 32296163) with decreased rate of heterozygous SNVs and increased rate of homozygous SNVs are candidate AOHs.
Limitations: FetalExome does not exclude all chromosome anomalies such as trinucleotide repeat expansions, triploidy, and mosaicism of SNV/InDel. Mutations in some genes cannot be detected due to technical reasons including local sequence characteristics or the presence of closely related pseudogenes.
Turn Around Time (TAT): 14 calendar days (Trio), 28 calendar days (Proband only).
Specimen requirement: Please contact our lab at (852) 3505 1557.
FetalExome Leaflet ( 繁中 / 簡中 )